Multi-Modal Surgery Pipeline with TOTALVI#
[1]:
import os
os.chdir('../')
import warnings
warnings.simplefilter(action='ignore', category=FutureWarning)
warnings.simplefilter(action='ignore', category=UserWarning)
[2]:
import scanpy as sc
import anndata
import torch
import scarches as sca
import matplotlib.pyplot as plt
import numpy as np
import scvi as scv
import pandas as pd
[3]:
sc.settings.set_figure_params(dpi=200, frameon=False)
sc.set_figure_params(dpi=200)
sc.set_figure_params(figsize=(4, 4))
torch.set_printoptions(precision=3, sci_mode=False, edgeitems=7)
Data loading and preprocessing#
For totalVI, we will treat two CITE-seq PBMC datasets from 10X Genomics as the reference. These datasets were already filtered for outliers like doublets, as described in the totalVI manuscript. There are 14 proteins in the reference.
[4]:
adata_ref = scv.data.pbmcs_10x_cite_seq(run_setup_anndata=False)
INFO Downloading file at data/pbmc_10k_protein_v3.h5ad
Downloading...: 24938it [00:00, 31923.71it/s]
INFO Downloading file at data/pbmc_5k_protein_v3.h5ad
Downloading...: 100%|█████████████████| 18295/18295.0 [00:05<00:00, 3198.56it/s]
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
[5]:
adata_query = scv.data.dataset_10x("pbmc_10k_v3")
adata_query.obs["batch"] = "PBMC 10k (RNA only)"
# put matrix of zeros for protein expression (considered missing)
pro_exp = adata_ref.obsm["protein_expression"]
data = np.zeros((adata_query.n_obs, pro_exp.shape[1]))
adata_query.obsm["protein_expression"] = pd.DataFrame(columns=pro_exp.columns, index=adata_query.obs_names, data = data)
INFO Downloading file at data/10X/pbmc_10k_v3/filtered_feature_bc_matrix.h5
Downloading...: 37492it [00:02, 13500.06it/s]
Variable names are not unique. To make them unique, call `.var_names_make_unique`.
Variable names are not unique. To make them unique, call `.var_names_make_unique`.
Now to concatenate the objects, which intersects the genes properly.
[6]:
adata_full = anndata.concat([adata_ref, adata_query])
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
And split them back up into reference and query (but now genes are properly aligned between objects).
[7]:
adata_ref = adata_full[np.logical_or(adata_full.obs.batch == "PBMC5k", adata_full.obs.batch == "PBMC10k")].copy()
adata_query = adata_full[adata_full.obs.batch == "PBMC 10k (RNA only)"].copy()
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
We run gene selection on the reference, because that’s all that will be avaialble to us at first.
[8]:
sc.pp.highly_variable_genes(
adata_ref,
n_top_genes=4000,
flavor="seurat_v3",
batch_key="batch",
subset=True,
)
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
Finally, we use these selected genes for the query dataset as well.
[9]:
adata_query = adata_query[:, adata_ref.var_names].copy()
Create TOTALVI model and train it on CITE-seq reference dataset#
[10]:
sca.models.TOTALVI.setup_anndata(
adata_ref,
batch_key="batch",
protein_expression_obsm_key="protein_expression"
)
INFO Using batches from adata.obs["batch"]
INFO No label_key inputted, assuming all cells have same label
INFO Using data from adata.X
INFO Computing library size prior per batch
INFO Using protein expression from adata.obsm['protein_expression']
INFO Using protein names from columns of adata.obsm['protein_expression']
INFO Successfully registered anndata object containing 10849 cells, 4000 vars, 2 batches,
1 labels, and 14 proteins. Also registered 0 extra categorical covariates and 0
extra continuous covariates.
INFO Please do not further modify adata until model is trained.
[11]:
arches_params = dict(
use_layer_norm="both",
use_batch_norm="none",
)
vae_ref = sca.models.TOTALVI(
adata_ref,
**arches_params
)
[12]:
vae_ref.train()
GPU available: True, used: True
TPU available: False, using: 0 TPU cores
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
Epoch 323/400: 81%|█▌| 323/400 [03:08<00:43, 1.76it/s, loss=1.23e+03, v_num=1]Epoch 323: reducing learning rate of group 0 to 2.4000e-03.
Epoch 358/400: 90%|█▊| 358/400 [03:28<00:24, 1.74it/s, loss=1.23e+03, v_num=1]Epoch 358: reducing learning rate of group 0 to 1.4400e-03.
Epoch 395/400: 99%|█▉| 395/400 [03:50<00:02, 1.71it/s, loss=1.22e+03, v_num=1]Epoch 395: reducing learning rate of group 0 to 8.6400e-04.
Epoch 400/400: 100%|██| 400/400 [03:53<00:00, 1.71it/s, loss=1.22e+03, v_num=1]
Save Latent representation and visualize RNA data#
[13]:
adata_ref.obsm["X_totalVI"] = vae_ref.get_latent_representation()
sc.pp.neighbors(adata_ref, use_rep="X_totalVI")
sc.tl.umap(adata_ref, min_dist=0.4)
[14]:
sc.pl.umap(
adata_ref,
color=["batch"],
frameon=False,
ncols=1,
title="Reference"
)
/home/marco/.pyenv/versions/scarches/lib/python3.7/site-packages/anndata/_core/anndata.py:1220: FutureWarning: The `inplace` parameter in pandas.Categorical.reorder_categories is deprecated and will be removed in a future version. Removing unused categories will always return a new Categorical object.
c.reorder_categories(natsorted(c.categories), inplace=True)
... storing 'batch' as categorical
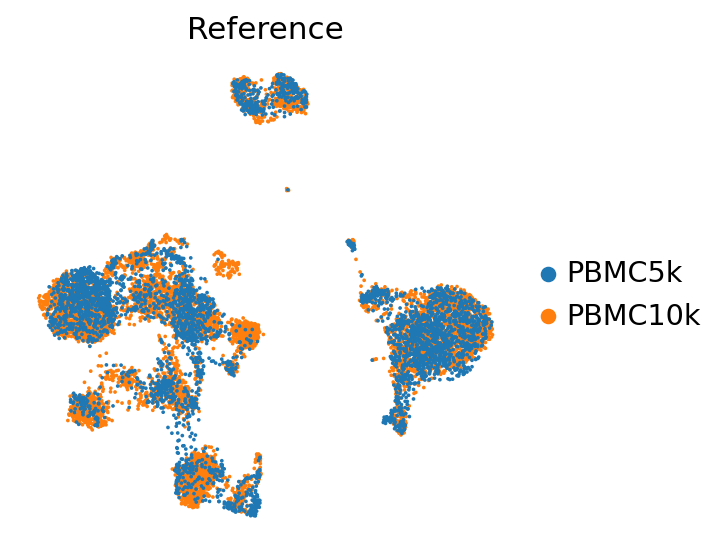
Save trained reference model#
[15]:
dir_path = "saved_model/"
vae_ref.save(dir_path, overwrite=True)
Perform surgery on reference model and train on query dataset without protein data#
[16]:
vae_q = sca.models.TOTALVI.load_query_data(
adata_query,
dir_path,
freeze_expression=True
)
INFO .obs[_scvi_labels] not found in target, assuming every cell is same category
INFO Found batches with missing protein expression
INFO Using data from adata.X
INFO Computing library size prior per batch
INFO Registered keys:['X', 'batch_indices', 'local_l_mean', 'local_l_var', 'labels',
'protein_expression']
INFO Successfully registered anndata object containing 11769 cells, 4000 vars, 3 batches,
1 labels, and 14 proteins. Also registered 0 extra categorical covariates and 0
extra continuous covariates.
/home/marco/.pyenv/versions/scarches/lib/python3.7/site-packages/scvi/model/base/_archesmixin.py:96: UserWarning: Query integration should be performed using models trained with version >= 0.8
"Query integration should be performed using models trained with version >= 0.8"
[17]:
vae_q.train(200, plan_kwargs=dict(weight_decay=0.0))
GPU available: True, used: True
TPU available: False, using: 0 TPU cores
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
Epoch 200/200: 100%|███████| 200/200 [02:56<00:00, 1.13it/s, loss=744, v_num=1]
[18]:
adata_query.obsm["X_totalVI"] = vae_q.get_latent_representation()
sc.pp.neighbors(adata_query, use_rep="X_totalVI")
sc.tl.umap(adata_query, min_dist=0.4)
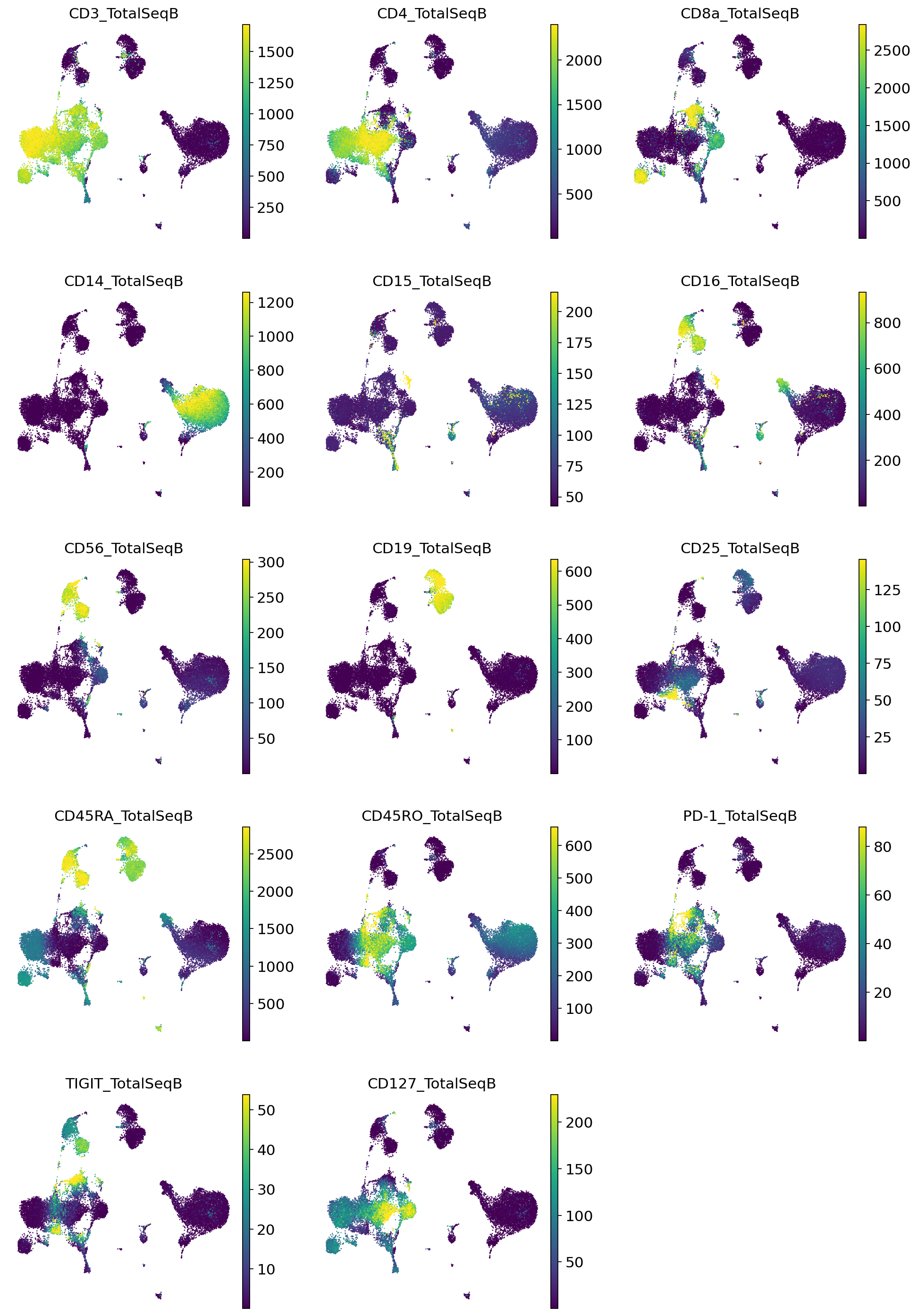
Impute protein data for the query dataset and visualize#
Impute the proteins that were observed in the reference, using the transform_batch parameter.
[19]:
_, imputed_proteins = vae_q.get_normalized_expression(
adata_query,
n_samples=25,
return_mean=True,
transform_batch=["PBMC10k", "PBMC5k"],
)
[20]:
adata_query.obs = pd.concat([adata_query.obs, imputed_proteins], axis=1)
sc.pl.umap(
adata_query,
color=imputed_proteins.columns,
frameon=False,
ncols=3,
)
/home/marco/.pyenv/versions/scarches/lib/python3.7/site-packages/anndata/_core/anndata.py:1220: FutureWarning: The `inplace` parameter in pandas.Categorical.reorder_categories is deprecated and will be removed in a future version. Removing unused categories will always return a new Categorical object.
c.reorder_categories(natsorted(c.categories), inplace=True)
... storing 'batch' as categorical
Get latent representation of reference + query dataset and compute UMAP#
[21]:
adata_full_new = adata_query.concatenate(adata_ref, batch_key="none")
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
[22]:
adata_full_new.obsm["X_totalVI"] = vae_q.get_latent_representation(adata_full_new)
sc.pp.neighbors(adata_full_new, use_rep="X_totalVI")
sc.tl.umap(adata_full_new, min_dist=0.3)
INFO Input adata not setup with scvi. attempting to transfer anndata setup
INFO Found batches with missing protein expression
INFO Using data from adata.X
INFO Computing library size prior per batch
INFO Registered keys:['X', 'batch_indices', 'local_l_mean', 'local_l_var', 'labels',
'protein_expression']
INFO Successfully registered anndata object containing 22618 cells, 4000 vars, 3 batches,
1 labels, and 14 proteins. Also registered 0 extra categorical covariates and 0
extra continuous covariates.
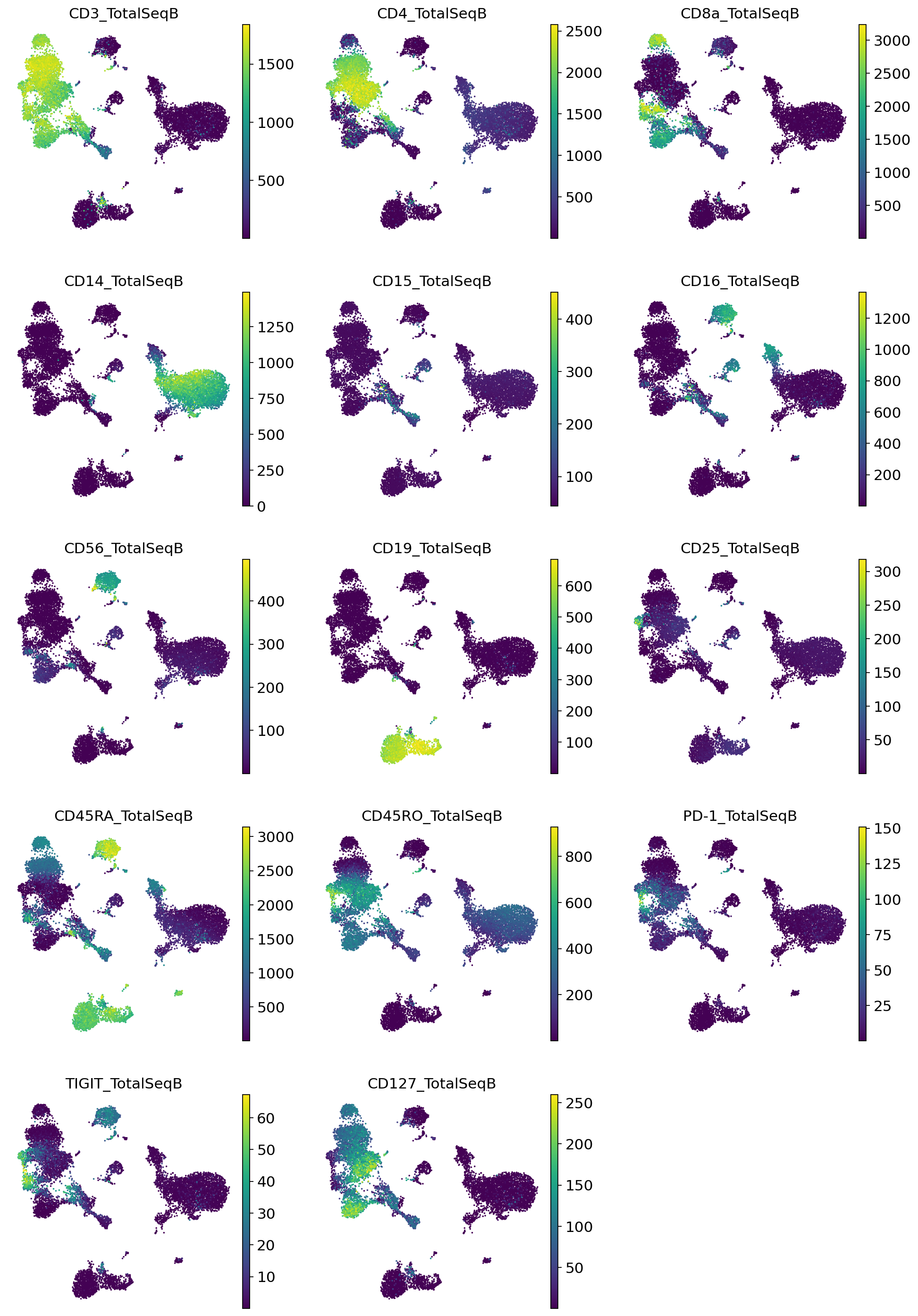
[23]:
_, imputed_proteins_all = vae_q.get_normalized_expression(
adata_full_new,
n_samples=25,
return_mean=True,
transform_batch=["PBMC10k", "PBMC5k"],
)
for i, p in enumerate(imputed_proteins_all.columns):
adata_full_new.obs[p] = imputed_proteins_all[p].to_numpy().copy()
[24]:
perm_inds = np.random.permutation(np.arange(adata_full_new.n_obs))
sc.pl.umap(
adata_full_new[perm_inds],
color=["batch"],
frameon=False,
ncols=1,
title="Reference and query"
)
/home/marco/.pyenv/versions/scarches/lib/python3.7/site-packages/anndata/_core/anndata.py:1220: FutureWarning: The `inplace` parameter in pandas.Categorical.reorder_categories is deprecated and will be removed in a future version. Removing unused categories will always return a new Categorical object.
c.reorder_categories(natsorted(c.categories), inplace=True)
/home/marco/.pyenv/versions/scarches/lib/python3.7/site-packages/anndata/_core/anndata.py:1229: ImplicitModificationWarning: Initializing view as actual.
"Initializing view as actual.", ImplicitModificationWarning
Trying to set attribute `.obs` of view, copying.
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
... storing 'batch' as categorical
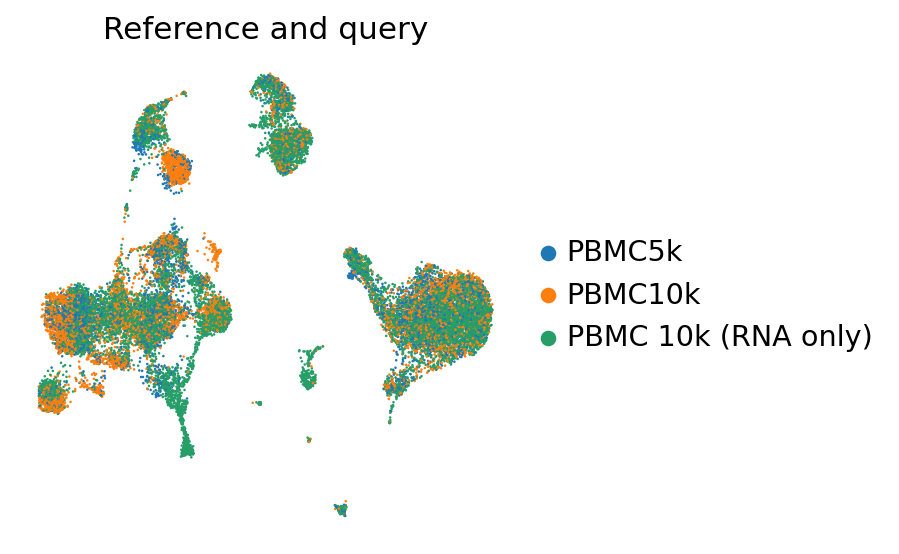
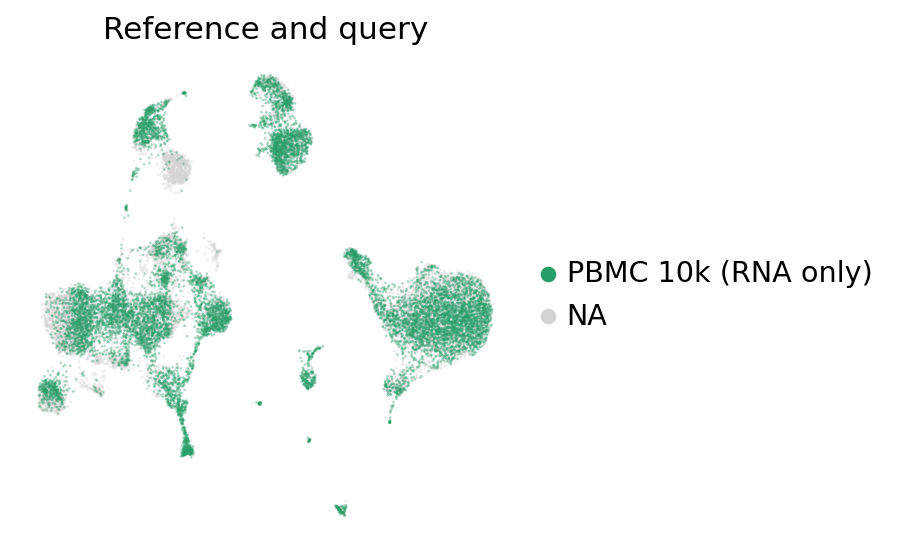
[25]:
ax = sc.pl.umap(
adata_full_new,
color="batch",
groups=["PBMC 10k (RNA only)"],
frameon=False,
ncols=1,
title="Reference and query",
alpha=0.4
)
/home/marco/.pyenv/versions/scarches/lib/python3.7/site-packages/anndata/_core/anndata.py:1220: FutureWarning: The `inplace` parameter in pandas.Categorical.reorder_categories is deprecated and will be removed in a future version. Removing unused categories will always return a new Categorical object.
c.reorder_categories(natsorted(c.categories), inplace=True)
... storing 'batch' as categorical
[26]:
sc.pl.umap(
adata_full_new,
color=imputed_proteins_all.columns,
frameon=False,
ncols=3,
vmax="p99"
)