Unsupervised surgery pipeline with SCVI
[1]:
import os
os.chdir('../')
import warnings
warnings.simplefilter(action='ignore', category=FutureWarning)
warnings.simplefilter(action='ignore', category=UserWarning)
[2]:
import scanpy as sc
import torch
import scarches as sca
from scarches.dataset.trvae.data_handling import remove_sparsity
import matplotlib.pyplot as plt
import numpy as np
import gdown
[3]:
sc.settings.set_figure_params(dpi=200, frameon=False)
sc.set_figure_params(dpi=200)
sc.set_figure_params(figsize=(4, 4))
torch.set_printoptions(precision=3, sci_mode=False, edgeitems=7)
Set relevant anndata.obs labels and training length
Here we use the CelSeq2 and SS2 studies as query data and the other 3 studies as reference atlas.
[4]:
condition_key = 'study'
cell_type_key = 'cell_type'
target_conditions = ['Pancreas CelSeq2', 'Pancreas SS2']
Download Dataset and split into reference dataset and query dataset
[5]:
url = 'https://drive.google.com/uc?id=1ehxgfHTsMZXy6YzlFKGJOsBKQ5rrvMnd'
output = 'pancreas.h5ad'
gdown.download(url, output, quiet=False)
Downloading...
From: https://drive.google.com/uc?id=1ehxgfHTsMZXy6YzlFKGJOsBKQ5rrvMnd
To: /home/marco/Documents/git_repos/scarches/pancreas.h5ad
126MB [00:03, 32.0MB/s]
[5]:
'pancreas.h5ad'
[6]:
adata_all = sc.read('pancreas.h5ad')
This line makes sure that count data is in the adata.X. Remember that count data in adata.X is necessary when using “nb” or “zinb” loss.
[7]:
adata = adata_all.raw.to_adata()
adata = remove_sparsity(adata)
source_adata = adata[~adata.obs[condition_key].isin(target_conditions)].copy()
target_adata = adata[adata.obs[condition_key].isin(target_conditions)].copy()
[8]:
source_adata
[8]:
AnnData object with n_obs × n_vars = 10294 × 1000
obs: 'batch', 'study', 'cell_type', 'size_factors'
[9]:
target_adata
[9]:
AnnData object with n_obs × n_vars = 5387 × 1000
obs: 'batch', 'study', 'cell_type', 'size_factors'
Create SCVI model and train it on reference dataset
Preprocess reference dataset. Remember that the adata file has to have count data in adata.X for SCVI/SCANVI if not further specified
[10]:
sca.models.SCVI.setup_anndata(source_adata, batch_key=condition_key)
INFO Using batches from adata.obs["study"]
INFO No label_key inputted, assuming all cells have same label
INFO Using data from adata.X
INFO Computing library size prior per batch
INFO Successfully registered anndata object containing 10294 cells, 1000 vars, 3 batches,
1 labels, and 0 proteins. Also registered 0 extra categorical covariates and 0 extra
continuous covariates.
INFO Please do not further modify adata until model is trained.
Create the SCVI model instance with ZINB loss as default. Insert “gene_likelihood=’nb’,” to change the reconstruction loss to NB loss.
[11]:
vae = sca.models.SCVI(
source_adata,
n_layers=2,
encode_covariates=True,
deeply_inject_covariates=False,
use_layer_norm="both",
use_batch_norm="none",
)
[12]:
vae.train()
GPU available: True, used: True
TPU available: False, using: 0 TPU cores
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
Epoch 400/400: 100%|███████| 400/400 [03:31<00:00, 1.89it/s, loss=502, v_num=1]
Create anndata file of latent representation and compute UMAP
[13]:
reference_latent = sc.AnnData(vae.get_latent_representation())
reference_latent.obs["cell_type"] = source_adata.obs[cell_type_key].tolist()
reference_latent.obs["batch"] = source_adata.obs[condition_key].tolist()
[14]:
sc.pp.neighbors(reference_latent, n_neighbors=8)
sc.tl.leiden(reference_latent)
sc.tl.umap(reference_latent)
sc.pl.umap(reference_latent,
color=['batch', 'cell_type'],
frameon=False,
wspace=0.6,
)
... storing 'cell_type' as categorical
... storing 'batch' as categorical

After pretraining the model can be saved for later use
[15]:
ref_path = 'ref_model/'
vae.save(ref_path, overwrite=True)
Perform surgery on reference model and train on query dataset
[16]:
model = sca.models.SCVI.load_query_data(
target_adata,
ref_path,
freeze_dropout = True,
)
INFO .obs[_scvi_labels] not found in target, assuming every cell is same category
INFO Using data from adata.X
INFO Computing library size prior per batch
INFO Registered keys:['X', 'batch_indices', 'local_l_mean', 'local_l_var', 'labels']
INFO Successfully registered anndata object containing 5387 cells, 1000 vars, 5 batches,
1 labels, and 0 proteins. Also registered 0 extra categorical covariates and 0 extra
continuous covariates.
[17]:
model.train(max_epochs=200, plan_kwargs=dict(weight_decay=0.0))
GPU available: True, used: True
TPU available: False, using: 0 TPU cores
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
Epoch 200/200: 100%|██| 200/200 [00:45<00:00, 4.37it/s, loss=1.16e+03, v_num=1]
[18]:
query_latent = sc.AnnData(model.get_latent_representation())
query_latent.obs['cell_type'] = target_adata.obs[cell_type_key].tolist()
query_latent.obs['batch'] = target_adata.obs[condition_key].tolist()
[19]:
sc.pp.neighbors(query_latent)
sc.tl.leiden(query_latent)
sc.tl.umap(query_latent)
plt.figure()
sc.pl.umap(
query_latent,
color=["batch", "cell_type"],
frameon=False,
wspace=0.6,
)
... storing 'cell_type' as categorical
... storing 'batch' as categorical
<Figure size 320x320 with 0 Axes>
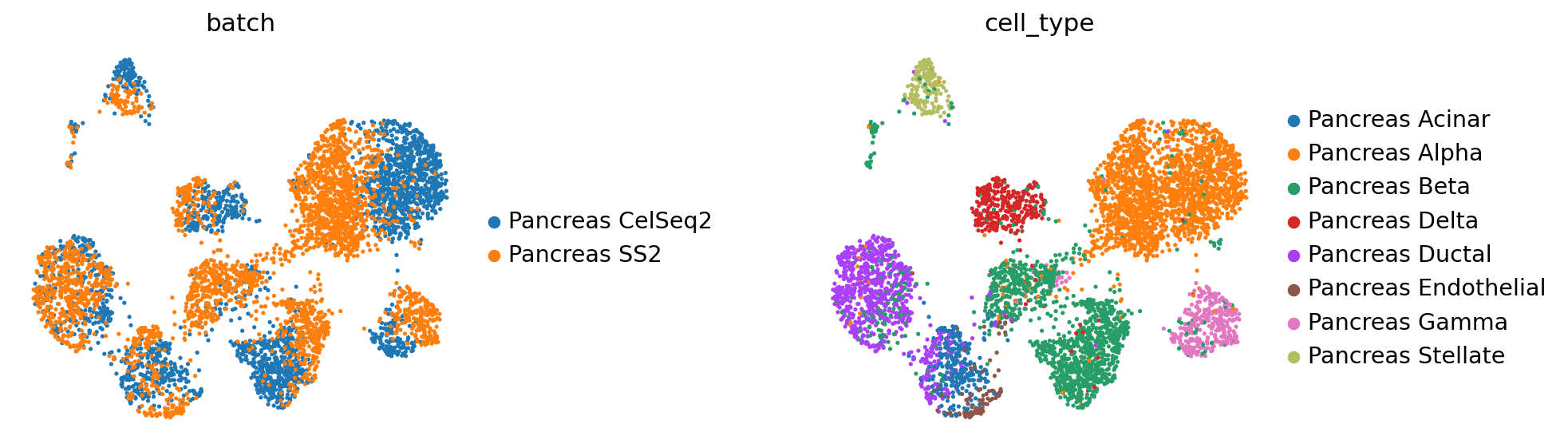
[20]:
surgery_path = 'surgery_model'
model.save(surgery_path, overwrite=True)
Get latent representation of reference + query dataset and compute UMAP
[21]:
adata_full = source_adata.concatenate(target_adata)
full_latent = sc.AnnData(model.get_latent_representation(adata=adata_full))
full_latent.obs['cell_type'] = adata_full.obs[cell_type_key].tolist()
full_latent.obs['batch'] = adata_full.obs[condition_key].tolist()
INFO Input adata not setup with scvi. attempting to transfer anndata setup
INFO Using data from adata.X
INFO Computing library size prior per batch
INFO Registered keys:['X', 'batch_indices', 'local_l_mean', 'local_l_var', 'labels']
INFO Successfully registered anndata object containing 15681 cells, 1000 vars, 5 batches,
1 labels, and 0 proteins. Also registered 0 extra categorical covariates and 0 extra
continuous covariates.
[22]:
sc.pp.neighbors(full_latent)
sc.tl.leiden(full_latent)
sc.tl.umap(full_latent)
plt.figure()
sc.pl.umap(
full_latent,
color=["batch", "cell_type"],
frameon=False,
wspace=0.6,
)
... storing 'cell_type' as categorical
... storing 'batch' as categorical
<Figure size 320x320 with 0 Axes>

[ ]: